1. Description of the methods¶
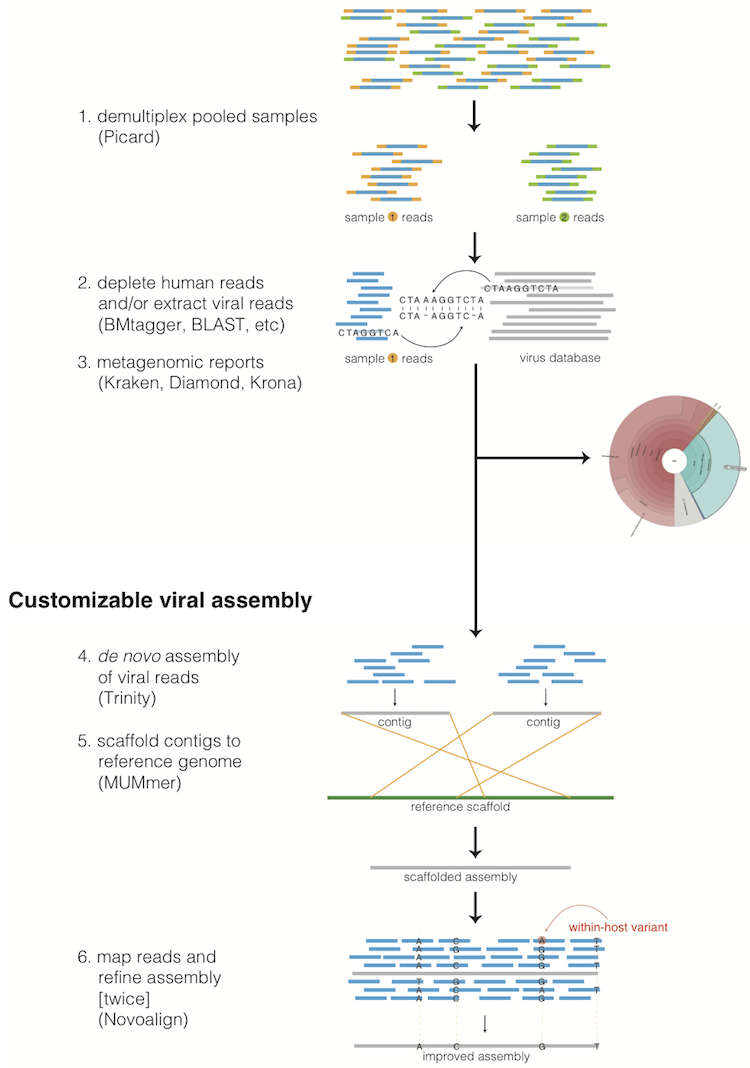
1.1. Taxonomic read filtration¶
1.1.1. Human, contaminant, and duplicate read removal¶
The assembly pipeline begins by depleting paired-end reads from each sample of human and other contaminants using BMTAGGER and BLASTN, and removing PCR duplicates using M-Vicuna (a custom version of Vicuna).
1.1.2. Taxonomic selection¶
Reads are then filtered to to a genus-level database using LASTAL, quality-trimmed with Trimmomatic, and further deduplicated with PRINSEQ.
1.2. Viral genome analysis¶
1.2.1. Viral genome assembly¶
The filtered and trimmed reads are subsampled to at most 100,000 pairs. de novo assemby is performed using Trinity. Reference-assisted assembly improvements follow (contig scaffolding, orienting, etc.) with MUMMER and MAFFT.
Each sample’s reads are aligned to its de novo assembly using Novoalign and any remaining duplicates were removed using Picard MarkDuplicates. Variant positions in each assembly were identified using GATK IndelRealigner and UnifiedGenotyper on the read alignments. The assembly was refined to represent the major allele at each variant site, and any positions supported by fewer than three reads were changed to N.
This align-call-refine cycle is iterated twice, to minimize reference bias in the assembly.
1.2.2. Intrahost variant identification¶
Intrahost variants (iSNVs) were called from each sample’s read alignments using V-Phaser2 and subjected to an initial set of filters: variant calls with fewer than five forward or reverse reads or more than a 10-fold strand bias were eliminated. iSNVs were also removed if there was more than a five-fold difference between the strand bias of the variant call and the strand bias of the reference call. Variant calls that passed these filters were additionally subjected to a 0.5% frequency filter. The final list of iSNVs contains only variant calls that passed all filters in two separate library preparations. These files infer 100% allele frequencies for all samples at an iSNV position where there was no intra-host variation within the sample, but a clear consensus call during assembly. Annotations are computed with snpEff.
1.3. Taxonomic read identification¶
Nothing here at the moment. That comes later, but we will later integrate it when it’s ready.
1.4. Cloud compute implementation¶
This assembly pipeline is also available via the DNAnexus cloud platform. RNA paired-end reads from either HiSeq or MiSeq instruments can be securely uploaded in FASTQ or BAM format and processed through the pipeline using graphical and command-line interfaces. Instructions for the cloud analysis pipeline are available at https://github.com/dnanexus/viral-ngs/wiki